Génová terapia sa všeobecne definuje ako dodanie alebo manipulácia s genetickým materiálom, s cieľom liečiť alebo predchádzať ochoreniam.
Existujú 4 prístupy génovej terapie, pri ktorých môžeme nahradiť gén, pridať gén, inaktivovať gén, ktorý je príčinou ochorenia, alebo cielene ho zmeniť.
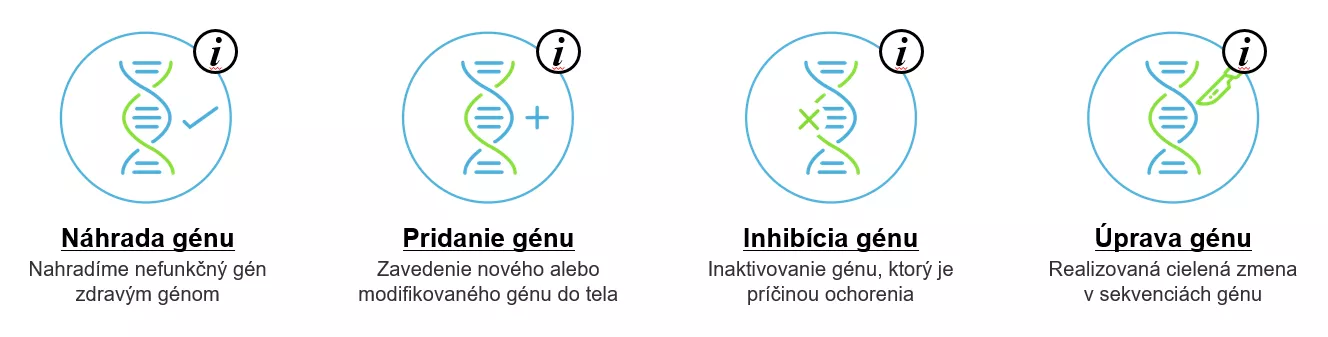
Jedným z ochorení, kde nahradením poškodeného génu funkčným génom vieme ovplyvniť priebeh ochorenia, je spinálna muskulárna atrofia.
Spinálna muskulárna atrofia (SMA) je autozomálne recesívne dedičné ochorenie, ktoré je charakterizované progresívnou degeneráciou motoneurónov a v tejto skupine dedičných ochorení patrí medzi najčastejšie a často smrteľné. Klinicky sa ochorenie prejavuje rastúcou progresívnou slabosťou a atrofizáciou priečne pruhovaného svalstva. Približne 1 z 54 ľudí je nositeľom tejto genetickej mutácie. SMA postihuje približne jedno z 10 000 živonarodených detí a je najčastejšou príčinou smrti u dojčiat zo všetkých genetických ochorení. V Európe sa ročne narodí 550 až 600 detí s touto diagnózou.
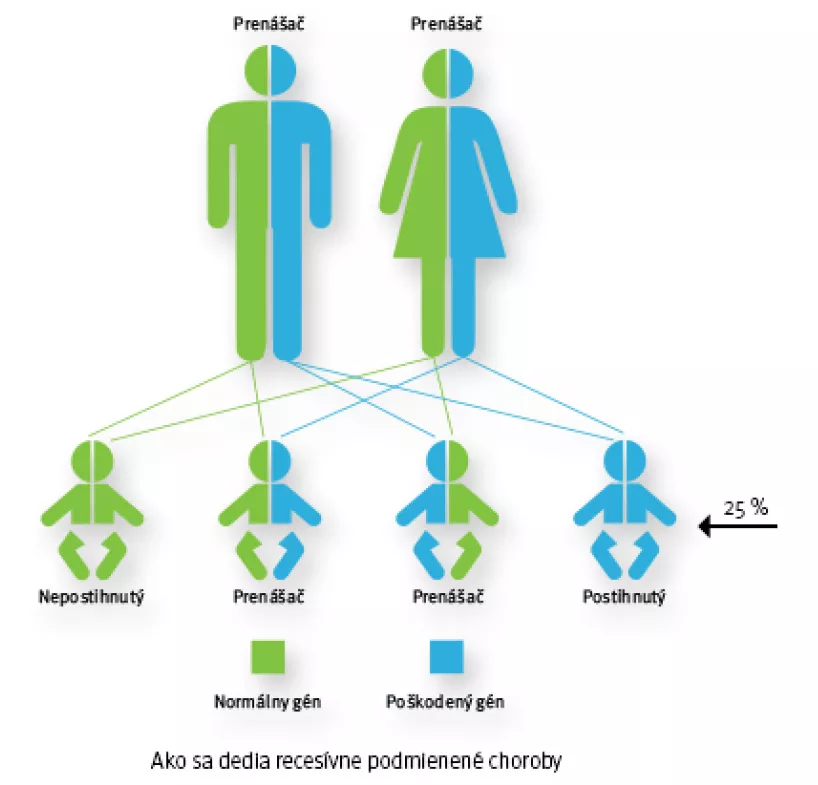
Pri recesívnej dedičnosti musí pacient zdediť 2 zmenené kópie určitého génu (t.j. od každého z rodičov dostane po jednej zmenenej kópii génu) na to, aby sa choroba prejavila. Ak človek zdedí len jeden zmutovaný gén a druhý je normálny, bude len zdravým prenášačom, pretože normálny gén funkčne kompenzuje mutovaný gén.

Ak sú obaja rodičia prenášačmi mutácie v rovnakom géne, môžu svojmu dieťatku odovzdať buď normálny alebo mutovaný gén. K prenosu dochádza úplne náhodne.
Každé dieťa, ktorého obaja rodičia sú prenášačmi mutácie v rovnakom géne, má riziko 25%, že mutáciu zdedí od oboch rodičov a choroba sa prejaví. U 75% sa ochorenie neprejaví a to buď preto, že zdedia len jednu kópiu mutovaného génu (50%) a sú zdraví prenášači, alebo zdedia normálne kópie génu a nie sú ani prenášačmi mutácie do ďalších generácií.
Príčinou spinálnej svalovej atrofie je mutácia génu, ktorý nazývame SMN 1 gén ( survival of motor neuron). U zdravých ľudí sa podľa zapísaných informácií v tomto géne tvorí proteín, ktorý zohráva zásadnú úlohu v prežívaní motoneurónov. Ak je tento gén mutovaný, pacienti nemajú alebo majú veľmi málo potrebného proteínu a dochádza k degenerácii motoneurónov.
Na základe veku, kedy sa ochorenie prejaví, závažnosti klinických príznakov, rýchlosti progresie ochorenia a prognózy, rozlišujeme niekoľko subtypov SMA:
- SMA I sa manifestuje do 6. mesiaca života a prežívanie bez liečby je maximálne do 2 rokov veku dieťatka.
- SMA II sa prejaví od 6. do 18. mesiaca veku a prežívanie je v rozsahu od 2 rokov až po rannú mladosť.
- SMA III sa môže prejaviť od 3 rokov do rannej mladosti a SMA IV väčšinou v 4. dekáde života. Oba typy SMA nemajú vplyv na dĺžku života človeka, avšak dramaticky znižujú jeho kvalitu.
Stupeň závažnosti postihnutia a rýchlosť progresie sa pri jednotlivých podtypoch líši, ale vo všeobecnosti k najzávažnejším symptómom patrí:
- znížený svalový tonus-hypotónia
- svalová slabosť priečne pruhovaného svalstva
- atrofia svalov
- areflexia
- zášklby
- problémy s dýchaním a prehĺtaním
- zmeny tvaru končatín, chrbtice a hrudníka v dôsledku slabosti svalov
- neschopnosť sedu a chôdze ( podľa typu SMA).
Mentálne postihnutie nepatrí k príznakom tohto ochorenia.

Spinálnu svalovú atrofiu diagnostikuje lekár väčšinou až na základe klinických príznakov. Je dôležité, aby sa liečba SMA začala čo najskôr a predišlo sa tak nezvratnej strate motoneurónov. Čím skôr sa liečba začne, tým je úspešnejšia. K včasnej diagnostike by mohlo významne prispieť zavedenie novorodeneckého skríningu a zahájenie liečby ešte v presymptomatickom období.